WebMO Basic Features
WebMO Basic is the FREE version of WebMO that includes an integrated 3-D molecular editor, interfaces to popular computational chemistry programs, built-in calculations, graphical visualization of results, external database connectivity, iPhone/Android compatibility, and web-based administration tools.
WebMO Basic Features
- Integrated 3D molecular editor and viewer
- Line structure editor for entering organic structures
- Support for most popular computational engines
- Built-in job queuing system and manager
- Integrated mechanics and Huckel MO calculations
- Graphical and tabular presentation of results
- Visualization of normal modes and IR, Raman, VCD, UV-VIS, NMR spectra
- Visualization of molecular symmetry elements
- Connection to many free external databases
- Phone/tablet support via the free WebMO app
- Web-based adminstration
- Compatible with all modern web browsers
Integrated 3D Molecular Editor and Viewer
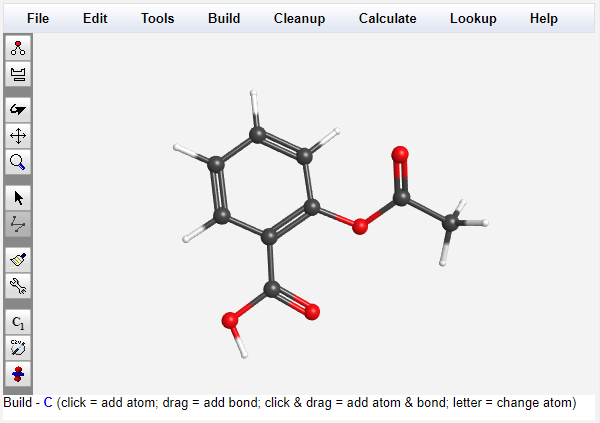
The built-in 3-D editor allows users to draw arbitrary molecules from an intuitive click-and-drag interface with support for rotation, translation, and zooming.
Clean-up automatically adds hydrogens and idealizes bond lengths and angles. Bond lengths, angles, and dihedrals may be displayed and adjusted. Fragments can be inserted, translated, and rotated.
Existing structures in XYZ, mol, or PDB format can be imported. The editor also allows for chemical information lookup, Huckel orbital calculations, and display of symmetry elements on structures.
Line Structure Editor
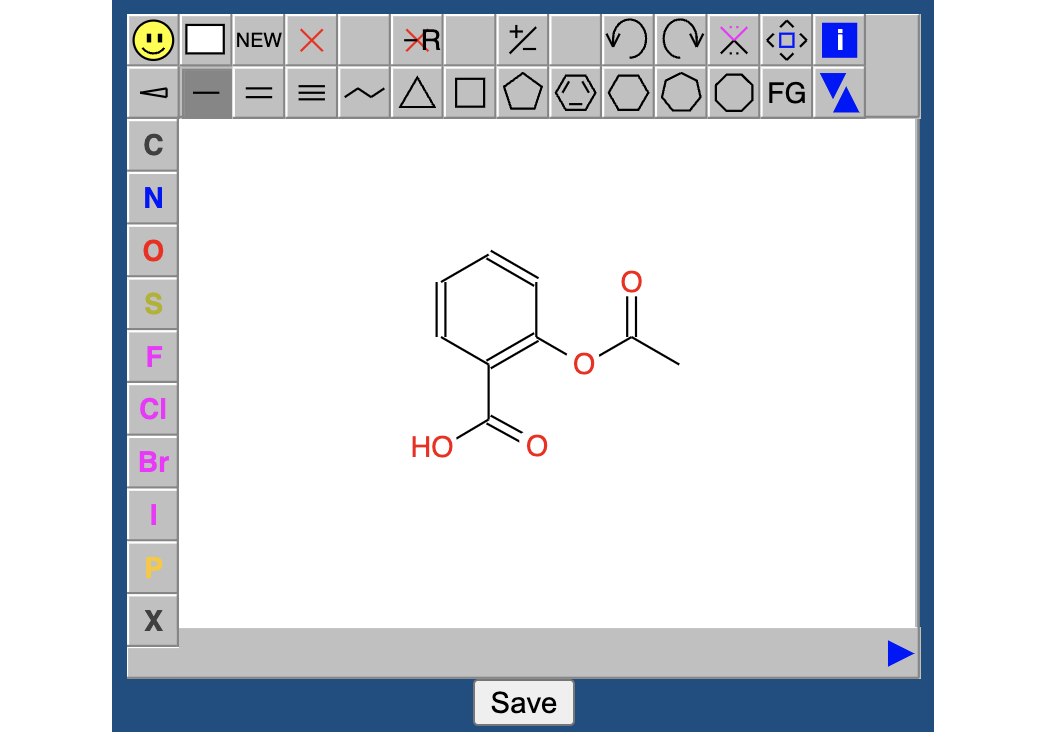
Alternatively, sketch molecules using the 2-D line structure editor, just as in organic chemistry.
2-D line structures are automatically converted into a 3-D when saved into WebMO. Stereochemistry is preserved during this process.
Support for Popular Computational Engines
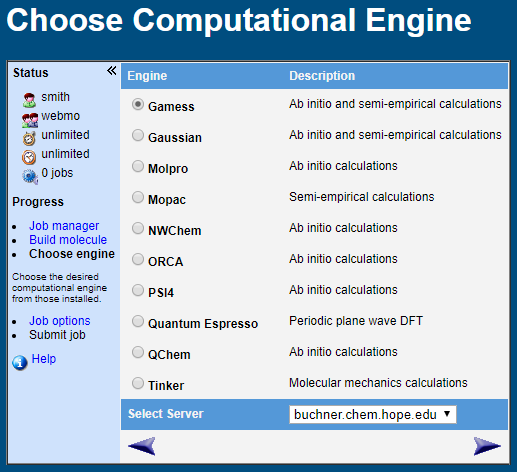
WebMO Basic supports most popular computational chemistry engines including: Gamess, Gaussian, MolPro, Mopac, NWChem, Orca, PQS, PSI, Q-Chem, TeraChem, Tinker, Quantum Expresso, VASP, and xTB
Supported calculation types include: molecular energy, geometry optimization, partial charges, bond orders, vibrational frequencies and spectra, thermochemistry, interaction energy, transition states, excited states, UV-Vis spectra, and NMR spectra
Built-In Job Queuing System and Manager
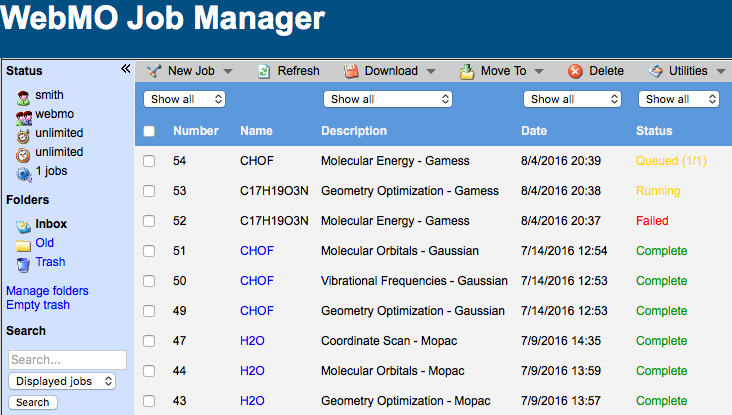
Submitted jobs are added to a built-in First In First Out (FIFO) queue.
The Job Manager displays all your computational chemistry jobs.
Completed, running, and queued jobs are listed. Completed jobs may be downloaded. Existing jobs from other sources may be imported.
Integrated Mechanics and Huckel MO Calculations
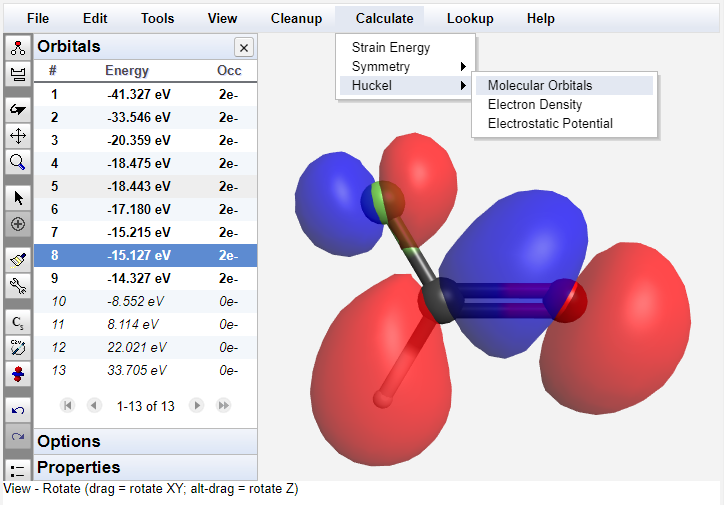
WebMO Basic includes built-in support for molecular mechanics calculations, symmetry analysis, and Huckel molecular orbital calculations.
These basic calculations are available without using a separate computational chemistry program.
Graphical and Tabular Presentation of Results
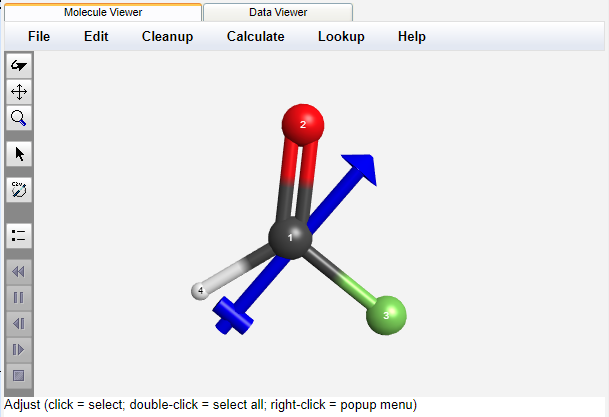
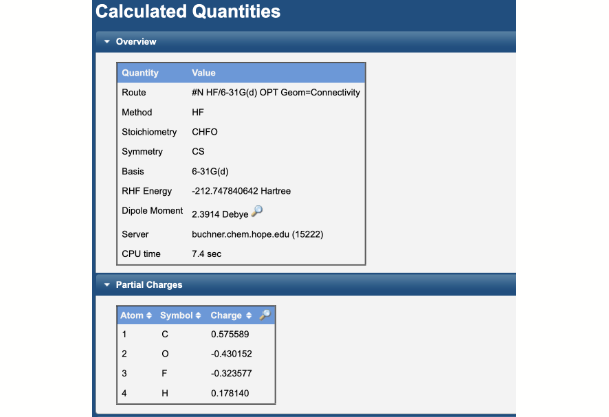
The 3-D viewer allows one to rotate, translate, and zoom the molecule, as well as display bond lengths, angles, and dihedrals. Partial charges and the dipole moment are visualized on the structure.
Tables of energies, thermochemistry results, rotational constants, vibrational frequencies, etc. are presented. The program raw output is also available.
Visualization of Normal Modes and Spectra
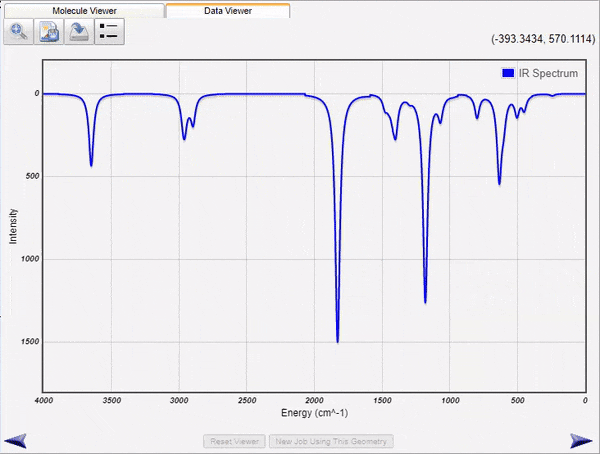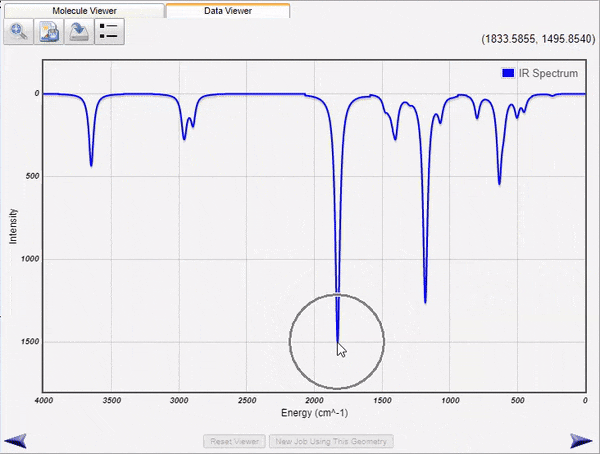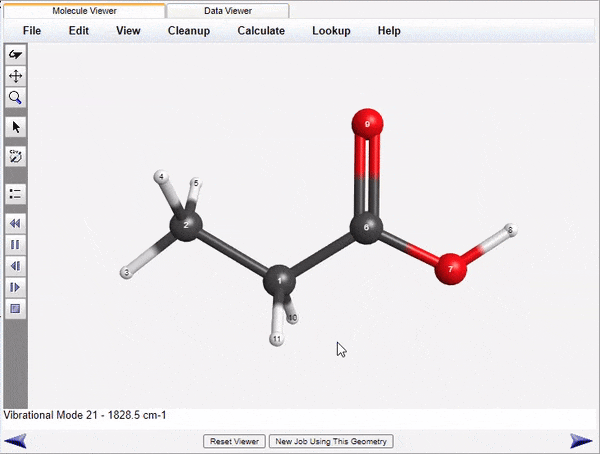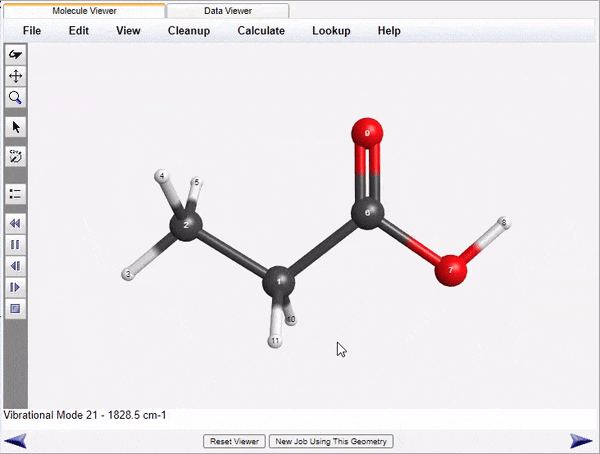
WebMO Basic run jobs to calculate infrared, Raman, VCD, UV-Vis, and NMR spectra of molecules. the results are parsed and displayed both as energy level tables and as spectra. Vibrational normal modes are display either with arrows to indicate atom motions or as animations.
The displayed spectra are linked to the molecular image. Clicking on an infrared peak will display its associated normal mode. Clicking on an NMR peak will highlight the associated atoms.
Realistic NMR spectra can be displayed using estimated or calculated proton splittings.
Visualize Symmetry Elements
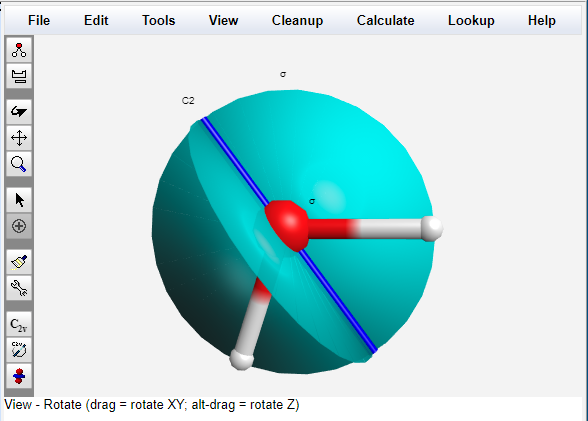
WebMO Basic can determine the point group of any molecule and display its symmetry elements, or any subset of symmetry elements.
A nearly symmetric molecule can also be symmetrized, which is useful prior to a calculation in order for the program to take advantage of the molecular symmetry.
Connection to Free External Databases
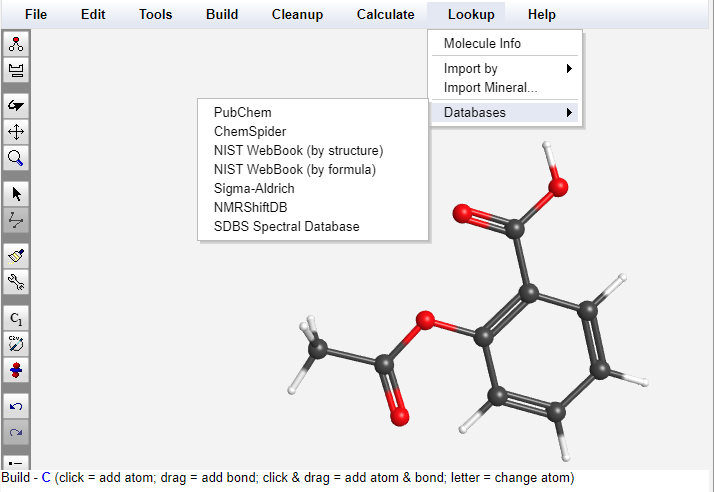
WebMO Basic can access and display results from a variety of external databases including PubChem, ChemSpider, NIST WebBook, Sigma-Aldrich, NMRShiftDB, and SDBS Spectral Database.
Once a molecule is imported or drawn, WebMO is a portal for displaying research, thermodynamic, safety, pricing, and spectral information available in these free databases.
Phone/Tablet Support via the Free WebMO App
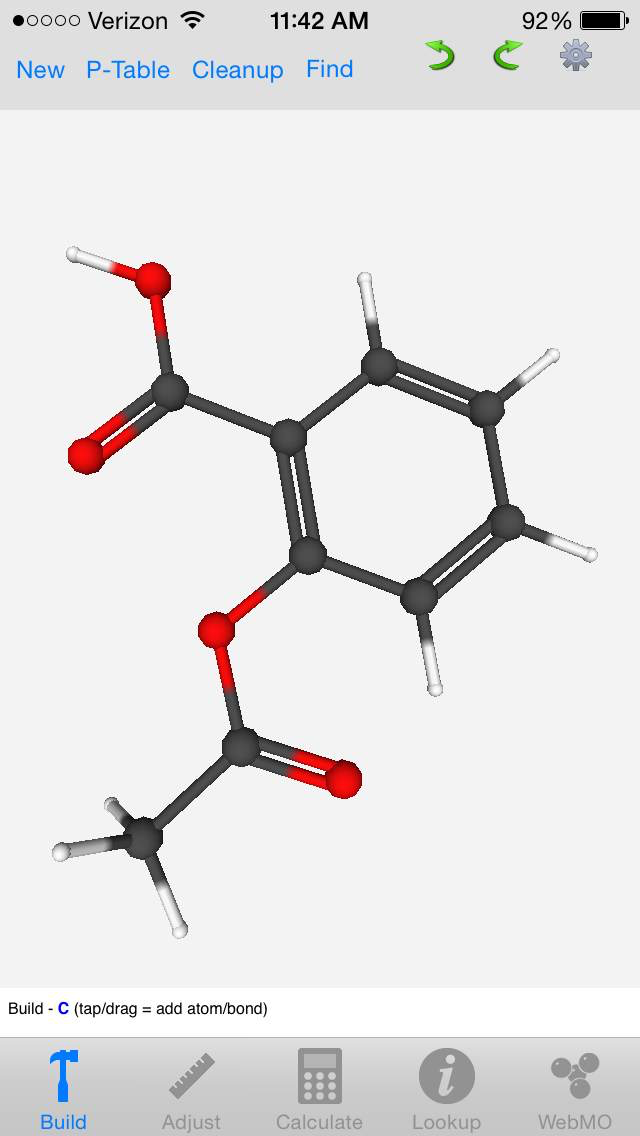
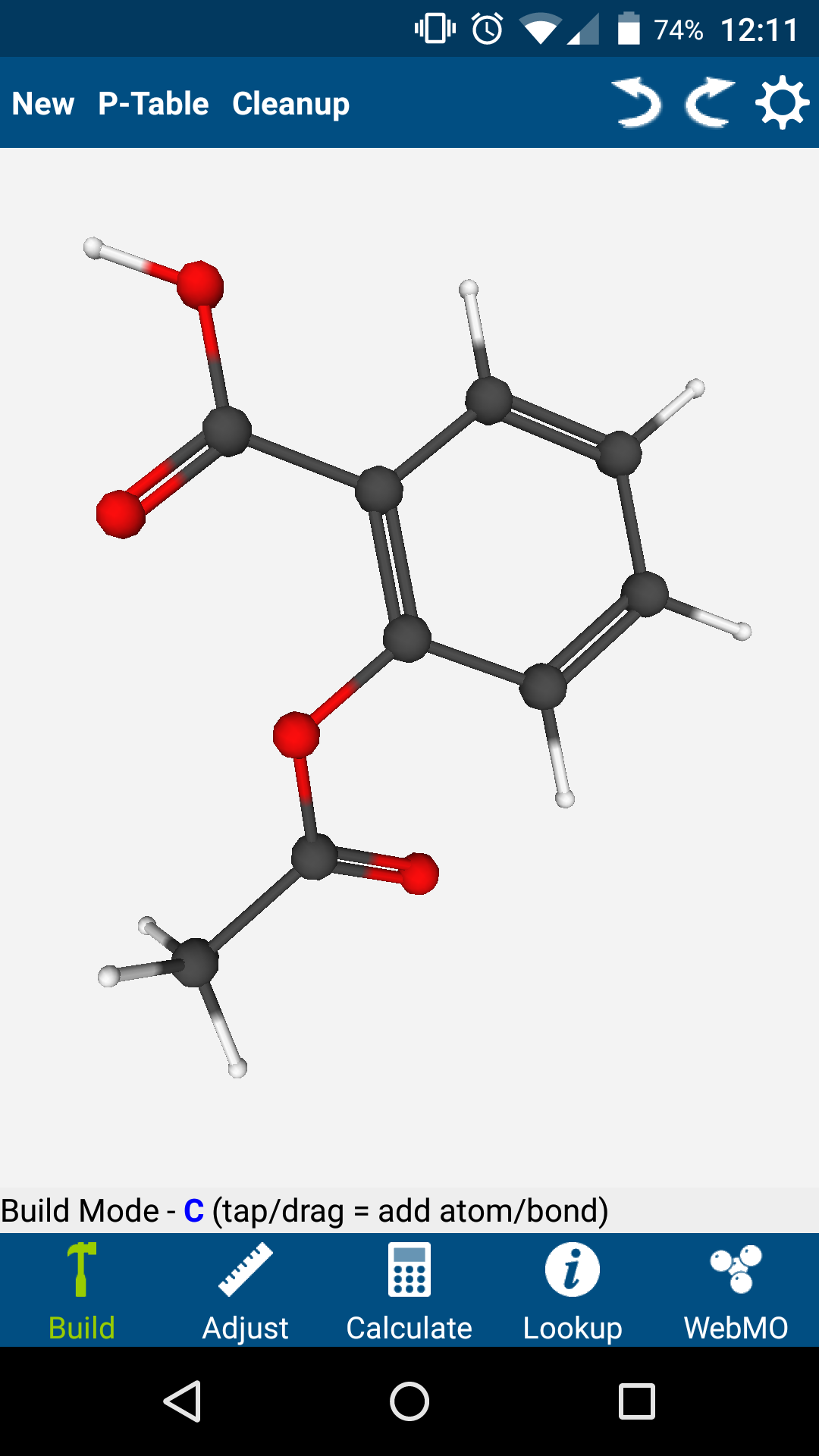
Support for Apple and Android phones and tablets is available using the free WebMO app.
Build molecules using an integrated editor, or import molecules by name. Lookup chemical information. Submit jobs to a WebMO server, and view calculated results.
Web-Based Adminstration
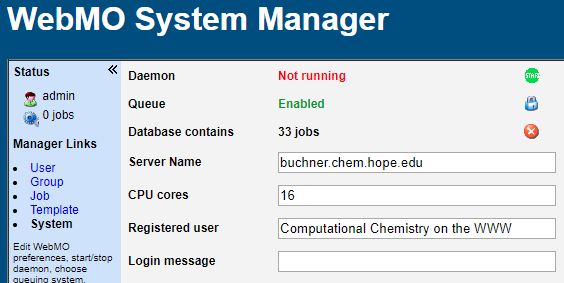
All WebMO administration is performed via a menu-based web interface.
Add, edit, disable, or delete WebMO users. View, download, or delete jobs from WebMO users. Limit CPU time on a per job or cumulative basis. Configure computational chemistry program settings. Configure system and WebMO settings.