Ab Initio Molecular Dynamics
Ab Initio Molecular Dynamics (AIMD) calculations model nuclear motion by numerically integrating Newton's equations of motion using the forces that arise from the potential energy surface determined by solving the Schrodinger Equation. The output of an AIMD calculation is a trajectory, which is the position of the atoms as a function of time, ie, the motion of the atoms.
AIMD calculations are computational intensive because the gradient of the energy must be computed at each time step in order to determine the force acting on each atom. The time steps are typically on the order of femtoseconds, while the length of a trajectory might be picoseconds. One must be judicious in the choice of step size and run length, as well as the choice of method and basis set size, to get reasonable run times.
There are two common modes for running AIMD trajectories:
- Constant Energy / NVE (microcanonical ensemble): Total energy is assumed to be constant, and energy is exchanged between potential and kinetic energy. This is like a ball rolling on a surface, except it is for the relative motion of all the atoms in the molecule.
- Constant Temperature / NVT (canonical ensemble): Kinetic energy is added or removed with a thermostat, so as to maintain constant temperature. This emulates other molecules colliding with the molecule so as to maintain constant kinetic energy, though these "collisions" can alter the directions of the constituent atoms. Various thermostat algorithms exist.
In addition, a choice must be made for the initial velocities of each atom. While the velocity of every atom could be individually specified, there are several common situations:
- Zero: Initially there is no kinetic energy, and the system evolves by going "downhill", keeping in mind that as kinetic energy accumulates it will affect the trajectory.
- Normal Mode: A small amount of kinetic energy is added to each atom in the direction of a normal mode. If the molecule is initially at equilibrium, this will result in normal mode vibrational motion. If the molecule is initially at a transition state, this will result in a Dynamic Reaction Coordinate (DRC), which is similar to an Intrinsic Reaction Coordinate (IRC) calculation except that kinetic energy is also included.
- Randomized Maxwell-Boltzmann distribution: Kinetic energy is randomly added to every atom in a manner consistent with a specific temperature. This is typically done for constant temperature as a good starting point. If done for constant energy, it is sometimes called "simulated annealing".
Specifying AIMD Conditions
After selecting Ab Initio Molecular Dynamics as the Calculation Type, the Advanced Job Options tab allows on to specify
- Step size, Number of steps or Run length, and Dump
- Constant E or Constant T
- Initial Velocities
Additional menu items are available for the specifying the vibrational mode / energy / direction (for a transition state) or temperature. The available options available depend on the capabilities of the engine.

AIMD Options on Advanced Tab
Visualizing AIMD Trajectories
The AIMD trajectory is available in the trajectory energy table. Clicking the view icon ( ) displays an interactive plot of kinetic, potential, and total energy. Clicking any point in this plot then displays the corresponding geometry.
) displays an interactive plot of kinetic, potential, and total energy. Clicking any point in this plot then displays the corresponding geometry.
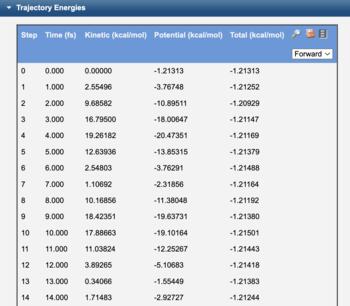 Trajectory Energy Table |
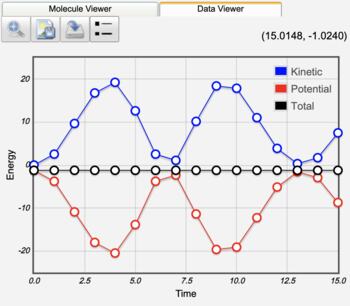 Interactive Trajectory Energy Plot |
Clicking the download icon ( ) allows one to download the trajectory in xyz format. Clicking the animate icon (
) allows one to download the trajectory in xyz format. Clicking the animate icon ( ) will display the trajectory as a movie. The direction (forward or reverse) and steps to be downloaded or animated may be specified.
) will display the trajectory as a movie. The direction (forward or reverse) and steps to be downloaded or animated may be specified.
In addition to the regular controls for viewing animations (rewind, play/pause, step backward, step forward, stop), the animation screen has a progress bar at the bottom which allows one to easily scroll through an animation to any step.
 H2CO Dissociation AIMD Trajectory |
 C2H6 500K Thermal Motion AIMD Trajectory |
Use screen capture software to create a *.mov file of the animation, which can then be edited and converted into an animated gif with free online tools such as ezgif.
Analyzing AIMD Trajectories and Animations
WebMO allows any two variables recorded in an animation to be plotted against each other. Furthermore, geometric parameters (bond distance, bond angle, dihedral angle) can be plotted along the trajectory.
In order to invoke the analysis tools, one must first display the animation by clicking the animate icon (). The Trajectory:Analysis... menu item will then be enabled. Selecting this menu item allows one to specify what is to be plotted on the X and (typically Step or Time) and on the Y axis (typically an Energy or geometric parameter). In order to select a geometric parameter (bond distance, bond angle, dihedral angle), 2, 3, or 4 atoms must have selected, respectively.
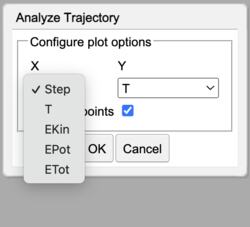 Analyze Trajectory X options |
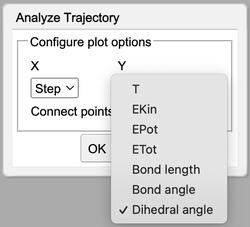 Analyze Trajectory Y options |
After selecting variables to be plotted on the X and Y axes, clicking OK displays the resulting analysis plot.
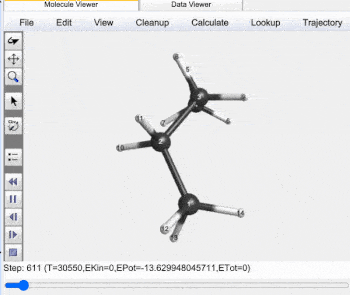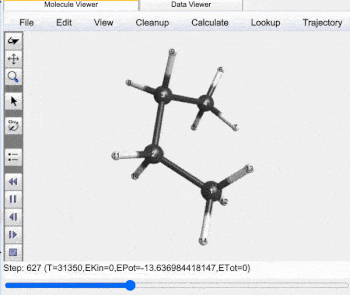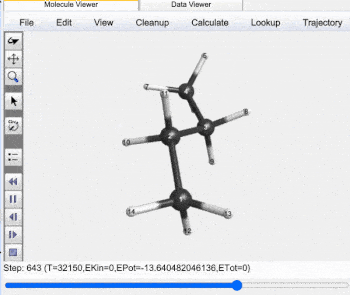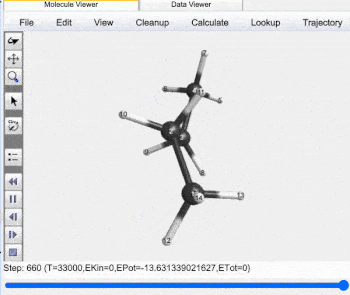 Butane Gauche to Anti Trajectory |
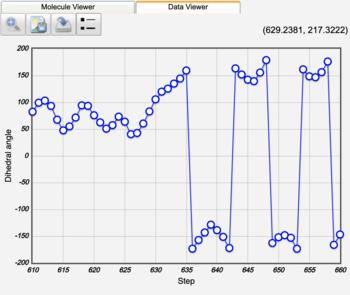 Gauche (60°) to Anti (±180°) Analysis |
The trajectory analysis tool is very general and can be used to analyze any animation, eg, geometry optimization, molecular vibration, reaction coordinate, AIMD trajectory, etc.