Configuring Job Options
The Configure Job Options page is where one specifies the details of the calculation to be carried out.
Frequently specified options (Name, Calculation, Theory, Basis) are listed the Job Options tab. Options that are less commonly used, specific to a particular program, or specific to a particular calculation type are specified on the Advanced tab.
One can also view and optionally the input file that is created with the selected options from the Preview tab.
It is also possible to add notes about the job with the Notes tab (WebMO Pro and Enterprise).
When done specifying options, click the forward arrow ( ) on the bottom right to submit the job to the WebMO job queue, where it is run in turn at the first available opportunity.
) on the bottom right to submit the job to the WebMO job queue, where it is run in turn at the first available opportunity.
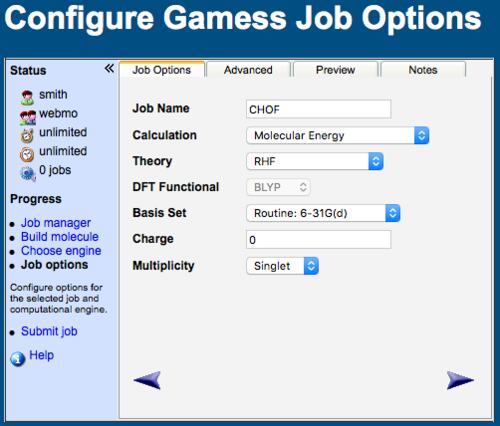
WebMO Configure Job Options Tab
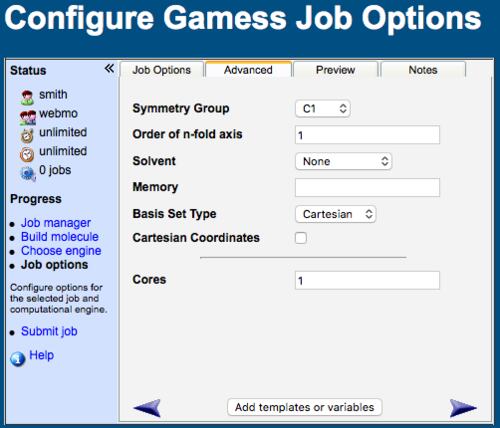
WebMO Advanced Job Options Tab
Common Job Options
Although reasonable defaults are provided for job options, one typically needs to modify the Job Name, Calculation, Theory, and Basis Set.
Job Name: The default is the molecular formula, but you will likely wish to specify a more descriptive title
Calculation: The default is Molecular Energy, but many other calculation types are available
Theory or Method: The default is usually Hartree-Fock, but many other theories (MP2, CCSD, DFT, etc.) are available
Functional: If a DFT method is selected, then a functional (B3LYP, MO5, etc.) must be specified
Basis Set: Minimal, Basic, Routine, and Accurate basis sets are suggested, but many others are listed; the basis set may also be specified with Other
Charge: The default is based on the overall charge of the molecule from the editor, but may be changed here
Multiplicity: The default is based on the number of electrons from the editor, but may be changed here
ONIOM: If layers exist, ONIOM method, charge, and multiplicity may be specified
Advanced Job Options
The advanced tab is individualized for each engine and calculation type. Although the following list is rather comprehensive, only the pertinent options for a selected calculation type are listed on the advanced tab.
Additional Keywords: specify additional keywords that are not provided by WebMO, e.g., "GEO-OK" allows atoms to be closer the 0.8 Angstroms in Mopac
Basis Set Type: Spherical has less linear dependence, but Cartesian are more easily implemented in the program; omitting a specification uses the program default
Calculate Force Constants: output force constants following a vibrational frequency calculation
Calculate Raman intensities: output Raman intensities
Cartesian Coordinates: specify the molecular geometry using cartesian coordinates instead of internal coordinates
Cores: Specify the number of CPU cores to be used in a calculation on a local or remote server (WebMO Pro and Enterprise); the more cores that are used, the faster the calculation
Density Fitting: Enable density fitting for pure DFT functions OR Calculate charges on each atom that reproduce the ESP (RESP)
Disable Symmetry: Turn off program optimization for molecular symmetry; useful when the point group of the molecule changes during the course of the calculation
EOM: Equation of Motion calculation for excited states
Excited State: Run the calculation for an excited states using a program-specific method
Excites State Method: Choose method for calculating the excited state
External Paramaters: Specify location of a file with external parameters
Freeze all core: Exclude core orbitals from post-SCF calculation
Include Connectivity: Include bonding information to assist program in determining internal coordinates
Integral Direct Mode: Calculate intergrals on-the-fly; do not pre-calculate or store them on disk for re-use
Max Opt Steps: Specify maximum number of steps during geometry optimization
Memory: Specify amount of memory allocated to job
Nodes : PPN : Node Type: Specify the number of compute nodes (typically 1), Processors (cores) Per Node (for example, 4), and compute node type (for example, bigmem) to be requested of the queueing system; this would be equivalent to a PBS line of "#PBS -l nodes=1:ppn=4:bigmem"
Optimization Coordinates: Specify coordinate system to be used for geometry optimization
Optimize Excited States: See Excited State
Output Mode: Choose Terse, Normal, or Verbose output
PCM Dielectric: See Solvent
Precise Optimization: Use tighter convergence criteria; useful for Frequency calculations
Resource List: Specify the resources to be requested of the queuing system (for example, walltime=24:00:00); this would be equivalent to a PBS line of "#PBS -l walltime=24:00:00"
Save Check Point/Restart File: Save a file from which the calculation can be restarted
Second Geometry: Specify the job number for a second geometry, e.g., in a Saddle calculation
Solvation Model: Specify solvent model
Solvent: Specify a solvent and use a program-specific continuous solvent model
Symmetry Group: Specify symmetry of molecule
Symmetry Number: Specify rotational symmetry number for statistical thermodynamic calculations
Use Check Point/Restart File: Specify job number of a calculation to be restarted